Ankara
Hafif Yağmurlu
11°C
Ankara
11°C
Hafif Yağmurlu
Pazartesi
Az Bulutlu
9°C
Salı
Çok Bulutlu
9°C
Çarşamba
Çok Bulutlu
9°C
Perşembe
Hafif Yağmurlu
8°C
Sun Savunma Net olarak bağımsız içeriklerimizi ücretsiz sunabilmek için reklam gelirlerine ihtiyaç duyuyoruz. Lütfen sitemizi desteklemek için reklam engelleyicinizi devre dışı bırakın ya da sitemizi beyaz listeye (whitelist) ekleyin.
Evlenirken test yaptırmayı unutmayın!
Akdeniz Anemisine Karşı Savaşta Kazanılan Çarpıcı Başarı


Akdeniz Anemisi (Talasemi) Türkiye’de en yaygın görülen genetik hastalıkların başında gelmektedir. Türkiye genelinde her 40-50 kişiden biri, Antalya, Adana ve Güneydoğu Anadolu Bölgesinde ise her 10 kişiden biri talasemi hastalığına yol açabilecek genleri taşımaktadır.
Yazar: Jerome Groopman, THE NEW YORKER, 18 Nisan 2018
Çeviren: Ercan Caner, Sun Savunma Net, 05 Eylül 2019
Yeni keşfedilen gen tedavisi, kırmızı kan hücrelerini etkileyen bir bozukluk olan beta talasemiyi yok edebilir. Foto: Shutterstock
1976’da tıp fakültesindeki son yılımda yurtdışına gittim ve İsrail’deki Hadassah Hastanesi hematoloji (kan bilimi) kliniğinde birkaç ay geçirdim. Orada bulunduğum süre boyunca her gün, beta talasemi olarak adlandırılan kan bozukluğundan etkilenen çocuklar ve gençlerle beraberdim.
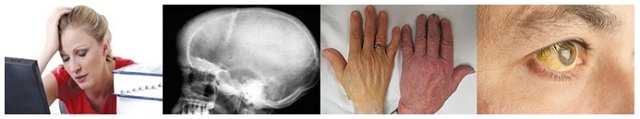
Kliniğin bekleme odasında onları ayırt etmek çok kolaydı. Tenleri soluk sarı renkte, kafatasları ve yüz kemikleri biçimsiz ve karın bölgeleri de genişlemiş karaciğer ve dalakları nedeniyle şişmiş durumdaydı. Birçoğu nefes darlığı, şişmiş bacaklar ve kalp yetmezliğinin çeşitli izlerini taşımaktaydılar.

Beta talasemi, daha iyi bilinen kuzeni orak hücreli anemi hastalığı gibi, kırmızı kan hücrelerimizin oksijen taşımasına imkân sağlayan hemoglobinin önemli bir bileşeni olan, globin proteininde doğuştan olan bir bozukluktan kaynaklanmaktadır. Sadece bir hatalı geni kalıtım yoluyla ebeveynlerinden alan çocuklarda genellikle hastalık semptomlarına rastlanmaz, iki hatalı geni alan çocuklarda ise hastalığın bütün etkileri görülür. Kırmızı kan hücreleri parçalanarak, etkilenen hastalarda ciddi anemiye neden olurlar.
Beta talasemi, her yıl tahmini olarak üç yüz bin insanı ve yeni doğan altmış bin çocuğu etkileyen dünyadaki en yaygın genetik hastalıklardan bir tanesidir. Beta talasemi hastalığı, Afrika, Asya, Orta Doğu ve özellikle Akdeniz bölgesinde yaygın olarak görülmektedir. Hastalığın adı Yunancada deniz anlamına gelen ‘‘thalassa’’ ve kan anlamına gelen ‘‘emia’’ sözcüklerinden gelmektedir. İngiltere, Yunanistan, İran, Suudi Arabistan ve Tayvan dâhil birçok ülkede çiftler, hastalığın belirlenmesi ve çocuklarına bu hastalığın kalıtım yoluyla aktarılma şansını öğrenebilsinler diye hamilelik öncesinde taramadan geçirilmektedir.
Bir hasta beta talasemi belirtileri gösterdiğinde esas olarak, sağlıklı vericilerden alınan düzenli kırmızı kan hücre naklinden ibaret palyatif tedavi yöntemi uygulanmaktadır. Fakat hayat kurtaran bu kan nakilleriyle büyük miktarda gelen demir, karaciğer, kalp ve diğer organlarda birikerek, hastalığın kendisinin verdiği zararı daha da büyütmektedir.
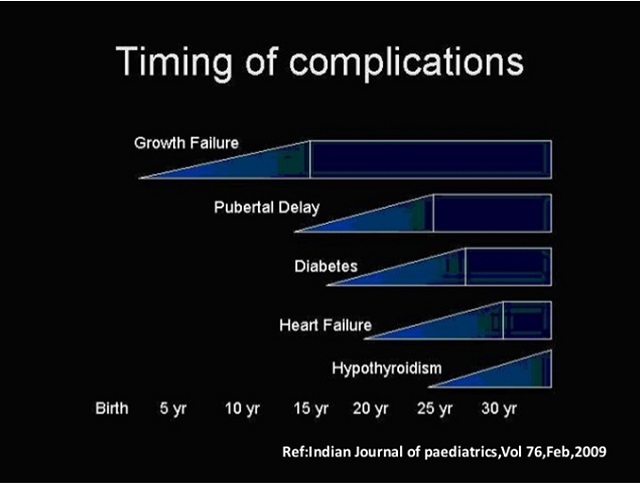
Talasemi Komplikasyonlarının Zamanlaması. Grafik: The New England Journal of Medicine. Growth Failure-Gelişme Bozukluğu, Pubertal Delay- Gecikmiş Ergenlik, Diabetes- Şeker Hastalığı, Hearth Failure- Kalp Yetmezliği, Hypothyroidism- Tiroit Yetmezliği. Birth- Doğum, Yr- Year- Yaş
Hadassah hastanesinde şiddetli beta talasemi formu rahatsızlığı olan hastalardan çok azı yetişkinliğe erişebilmiştir. Ölümlerinden önceki son yıllarda hastalarda, tipik olarak kemik kırılmaları, sürekli yinelenen enfeksiyonlar ve karşı koyulamayan yorgunluk belirtileri görülmektedir.
O genç hastalarla ilgilenmemin üzerinden kırk yıldan fazla zaman geçtikten sonra bilim, şimdi hastalığı tedavi etme aşamasına gelmiş durumdadır. Bu hafta, New England Journal of Medicine dergisinde, Birleşik Devletler, Fransa, Avustralya ve Tayland’dan araştırmacılar tarafından kaleme alınan, beta talasemi hakkında dönüm noktası niteliğinde bir makale yayınlanmıştır.
Beta talasemi hastalığından etkilenen ve dünyanın çeşitli yerlerinde bulunan altı merkezde tedavi altında olan yirmi iki hastaya, anormal genin yerini alacak olan, normal genin varyantının hastanın DNA’sına yerleştirildiği, gen terapisi adı verilen bir tedavi uygulanmıştır. Bu olayda araştırmacılar her bir hastanın, vücudun kan fabrikası olan kemik iliğinden olgunlaşmamış kök hücrelerini almış ve bunları laboratuvar ortamında izole etmişlerdir.
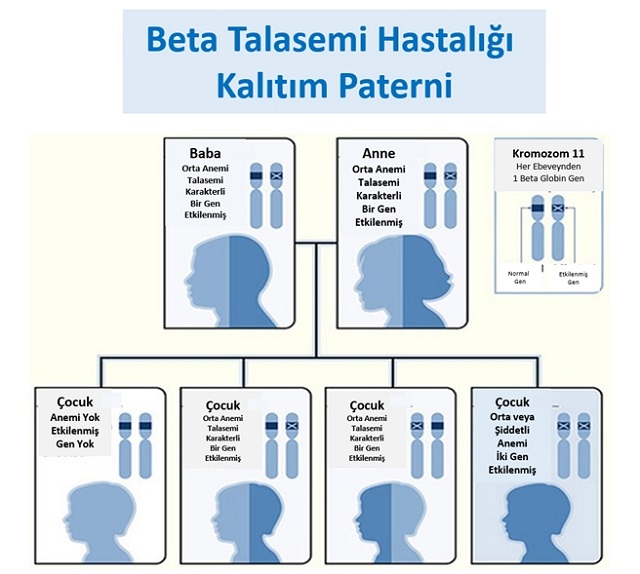
Beta Talasemi Kalıtım Paterni. İllüstrasyon: SERUM Thalassemia Prevention Federation
Sonra, hücrelere normal globin geninin kopyasını aşılamak maksadıyla zararsız bir virüs kullanmışlardır. Hastanın iliğini hastalıklı genlerden kemoterapi ile temizledikten sonra, genetik olarak değiştirilmiş hücreleri kan dolaşımı sistemine, otolog transplantasyon adı verilen bir yöntemle yeniden aktarmışlardır. Hastanın kan dolaşım sistemine enjekte edilen hücreler, iliğe ulaşma işini kendi başlarına gerçekleştirmektedirler.
Araştırmacıların umudu, modifiye edilmiş kök hücrelerin, kırmızı kan hücreleri içinde olgunluğa erişerek çok sayıda sağlıklı hemoglobin üretmeleridir. Umut gerçekleşir. Yirmi iki hastadan dokuzu ağır beta talasemi hastalığına maruzdur ve tedavi sonrasında bu hastaların ihtiyaç duydukları kan nakli sayısı %74 oranında azalmıştır. Dokuz hastadan üçü ise artık kan nakline hiç ihtiyaç duymamaktadır.
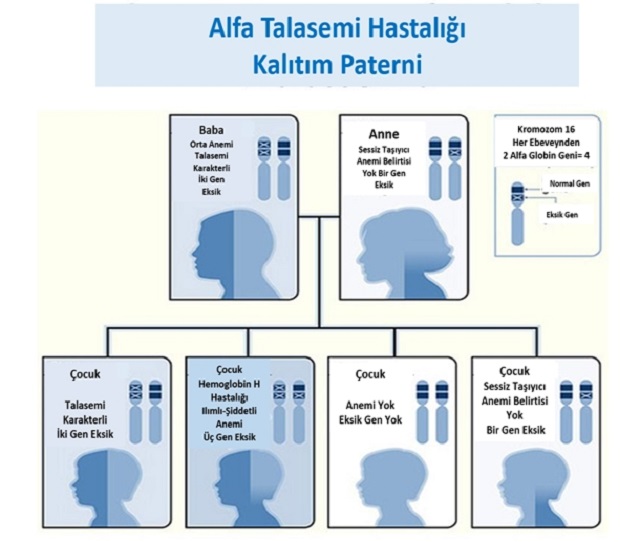
Alfa Talasemi Kalıtım Paterni. İllüstrasyon: SERUM Thalassemia Prevention Federation
Geri kalan, hastalığın daha hafifine yakalanan on üç hastadan on ikisi için de aynı şeyler geçerlidir. Bugüne kadar deneye katılan hastalar, azami kırk iki ay süreyle gözlenmiştir, fakat tedavinin faydalarının sürdüğünü ve yan etkileri olmadığını garanti etmek maksadıyla, hastalar gelecekte de izlenmeye devam edilecektir. Deneylerin erken safhalarında, prosedürün kök hücrelerin DNA’sını tahrip edeceği ve potansiyel olarak kan kanserini tetikleyeceği yönündeki endişeler neyse ki gerçekleşmemiştir.
Alınan sonuçlar gerçekten müthiştir. Bu dramatik başarının nedeni nedir? Bu türden bütün büyük başarılarda olduğu gibi bu araştırma ve deneylerin başarısı da paralel alanlarda onlarca yıl süren ve adım adım ilerleyen çalışmalara dayanmaktadır.
Kemik iliği naklinde ilk teşebbüsler, Harvard üniversitesinde eğitim gören E. Donnall Thomas adlı fizikçinin, köpekler üzerinde deneylere başladığı, 1950’li yılların ortalarına kadar gitmektedir. 1960’lı yılların sonlarına kadar uzanan ilk testler neredeyse değişmez bir şekilde hastaların ölümü ile sonuçlanmıştır. Takip eden yıllarda ise son derece travmatik olan bu prosedür (hâlâ travmatik olsa da) güzel bir sanata dönüşmüştür.
Gen tedavisi de uzun aşamalardan geçmiştir. Beta talasemi deneyinde, modifiye edilmiş globinin başarılı bir şekilde vücuda verilmesi, dergide yayınlanan makalenin yazarlarından Philippe Leboulch tarafından geliştirilen, doğal olarak ortaya çıkan ve hücreleri fark gözetmeksizin etkileyen bir patojen olan lentivirüs üzerine bindirilen, taşıyıcı veya dağıtma sistemi sayesinde mümkün olabilmiştir.
Makaleye eşlik eden bir başyazıda, Dana-Farber/Boston Çocuk Kanser ve Kan Bozuklukları Merkezinden Alessandra Biffi, bazı bağışıklık sistem yetmezliği olan ve hemofili hastalarını hedefleyen gen tedavilerinin son zamanlarda başarılı olduklarına dikkat çekmektedir.
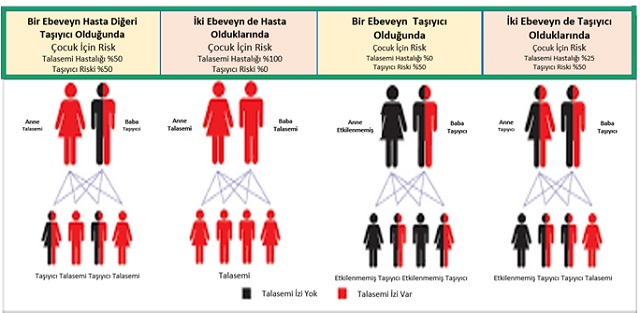
Şimdi halledilmesi gereken zorluk ise tıbbi tedavinin sınırlı olduğu dünyanın birçok yerinde karmaşık fakat hayat kurtaran bu tedavinin uygulanabilmesi için yöntemler bulmaktır. Otolog ilik nakli kurumları, tıpkı sağlıklı globin genlerini kök hücrelere aşılayacak laboratuvarlar gibi, dünyanın birçok gelişmiş ülkesinde mevcuttur. Fakat beta talaseminin en fazla yaygın olduğu dünyanın bazı yerlerinde, bu tesisler ne yazık ki hâlâ yoktur.
Bu tesisleri finanse etme ve yapmanın lehine kuvvetli insani bir argümanın yanı sıra ekonomik açıdan sorunlar da mevcuttur. Beta talasemi, tedavisi uzun vadeli ve inanılmaz ölçüde pahalı olan bir hastalıktır ve gen tedavisi de tek dozluk bir tedavi anlamına gelmektedir. Halsizlik ve erken ölümle göze çarpan, kalıtım yoluyla geçen bu öldürücü hastalık, bu hastalıktan etkilenenleri sağlığına kavuşturmak ve ölümleri engellemeyi bir öncelik haline getirme arzumuz olur ise tedavi edilebilir.
Aşağıda The New England Journal of Medicine dergisinde yayınlanan makalenin kısa özetini okuyabilirsiniz.
Geçmiş
Donör uygunluğu ve transplantasyonla ilgili riskler, kan nakline bağımlı beta talasemi hastalarında allojenik hematopoietik kök hücre transplantasyonunu sınırlandırmaktadır. Belirli bir beta globin (βA-T87Q) gen lentiviral transferinin, uzun süreli kırmızı hücre transfüzyolarının yerini alabileceğini tespit ettikten sonra, transfüzyon bağımlısı β-talasemi hastalarında böyle bir gen tedavisinin güvenliliğini ve etkinliğini değerlendirmek istedik.
Safha 1 ve Safha 2 olarak adlandırdığımız iki safhalı çalışmamızda, yaşları 12 ile 35 arasında değişen transfüzyon bağımlısı 22 beta talasemi hastasından, mobilize otolog CD34+ hücrelerini aldık ve bu canlı hücreleri (ex vivo), yetişkin hemoglobini (HbA), bir T87Q amino asit ikamesi (HbAT87Q) ile kodlayan LentiGlobin BB305 taşıyıcı yöntemiyle transfer ettik. Hücreler daha sonra, hastaların iliklerindeki busulfan iyileştirilmesi sonrasında tekrar hastaların kan dolaşım sistemine enjekte edildiler. Sonra olumsuz olayları, taşıyıcı entegrasyonunu ve replikasyona dayanıklı lentivirüsü izledik. Yaptığımız etkinlik değerlendirmeleri; total hemoglobin ve HbAT87Q seviyelerini, kan nakli gereksinmelerini ve ortalama taşıyıcı kopya numarasını kapsamıştır.
Gen modifiyeli hücrelerin enjeksiyonundan ortalama 26 ay (Aralık 15-42) sonra β0/β0 genotip olan 13 hastadan bir tanesi hariç hiç birinde kırmızı hücre nakline gerek kalmadığını gördük. HbAT87Q seviyeleri, desilitrede 3,4 ile 10,0 arasında ve total hemoglobin seviyeleri de desilitrede 8,2 ile 13,7 arasında değerler gösterdi. İncelenen hastalarda hemoglobin seviyeleri normal aralığa düşürülerek, diseritropoiez biyolojik işaretleyicilerin düzeltilmesi başarıldı. β0/β0 genotip olan dokuz hastada veya IVS1-110 mutasyonu iki kopyasında, yıllık ortalama transfüzyon hacmi %73 oranında azaldı ve üç hastada kırmızı kan hücre transfüzyonuna tamamen gerek kalmadığı görüldü. Tedaviyle bağlantılı olumsuz olaylara otolog kök hücre transplantasyonlarında tipik olarak rastlanmaktadır. Uygulanan tedavi esnasında taşıyıcı entegrasyonuna bağlı klonal dominans gözlenmedi.
BB305 taşıyıcı enjekte edilen otolog CD34+ gen tedavisinin, şiddetli beta talasemiden rahatsız 22 hastada, ürünle bağlantılı ciddi olumsuz etkiler yaratmadan, uzun vadeli kırmızı hücre nakli ihtiyacını azalttığı veya ortadan kaldırdığı görüldü.
Çevirenin Notları: Bu yazının maksadı bilgi vermek ve Akdeniz Anemisi (Talasemi) hastalığının tedavisinde, devrim niteliğindeki bir araştırma ve deneyin sonuçlarını okuyuculara iletmektir. Herhangi bir sağlık sorununda sayın okurların sağlık kuruluşlarına danışmaları tavsiye edilir. Yazının orijinal metnine aşağıdaki link üzerinden erişebilirsiniz.
Evlenirken test yaptırmayı unutmayın!
A Cure for One of the World’s Most Devastating Blood Diseases?
In 1976, my final year of medical school, I travelled abroad and spent several months working in the hematology clinic at Hadassah Hospital, in Jerusalem. Every day, I attended to children and teen-agers suffering from a blood disorder called beta thalassemia. They were easy to identify in the clinic waiting room.